Aberrant activation of Wnt/β-catenin pathway is a key driver of colorectal cancer (CRC) growth and of great therapeutic importance. In this study, we performed comprehensive CRISPR screens to interrogate the regulatory network of Wnt/β-catenin signaling in CRC cells. We found marked discrepancies between the artificial TOP reporter activity and β-catenin–mediated endogenous transcription and redundant roles of T cell factor/lymphoid enhancer factor transcription factors in transducing β-catenin signaling. Compiled functional genomic screens and network analysis revealed unique epigenetic regulators of β-catenin transcriptional output, including the histone lysine methyltransferase 2A oncoprotein (KMT2A/Mll1). Using an integrative epigenomic and transcriptional profiling approach, we show that KMT2A loss diminishes the binding of β-catenin to consensus DNA motifs and the transcription of β-catenin targets in CRC. These results suggest that KMT2A may be a promising target for CRCs and highlight the broader potential for exploiting epigenetic modulation as a therapeutic strategy for β-catenin–driven malignancies.
Colorectal cancer (CRC) represents one of the major malignancies and a leading cause of cancer-related death worldwide. Aberrant Wnt/β-catenin pathway plays a pivotal role in colon carcinogenesis (1). Cytoplasmic β-catenin is phosphorylated by a protein complex containing adenomatous polyposis coli (APC), AXIN1 or AXIN2, casein kinase 1α (CK1α), and glycogen synthase kinase-3β (GSK3β), leading to β-catenin destruction through ubiquitin-proteasome system. Wnt binding to the LDL receptor related protein 5/6 (LRP5/6)–frizzled receptors results in the disassembly of the β-catenin–destruction complex and consequent accumulation of β-catenin. β-Catenin then enters into the nucleus and binds to T cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors to initiate the transcription of β-catenin downstream targets (2).
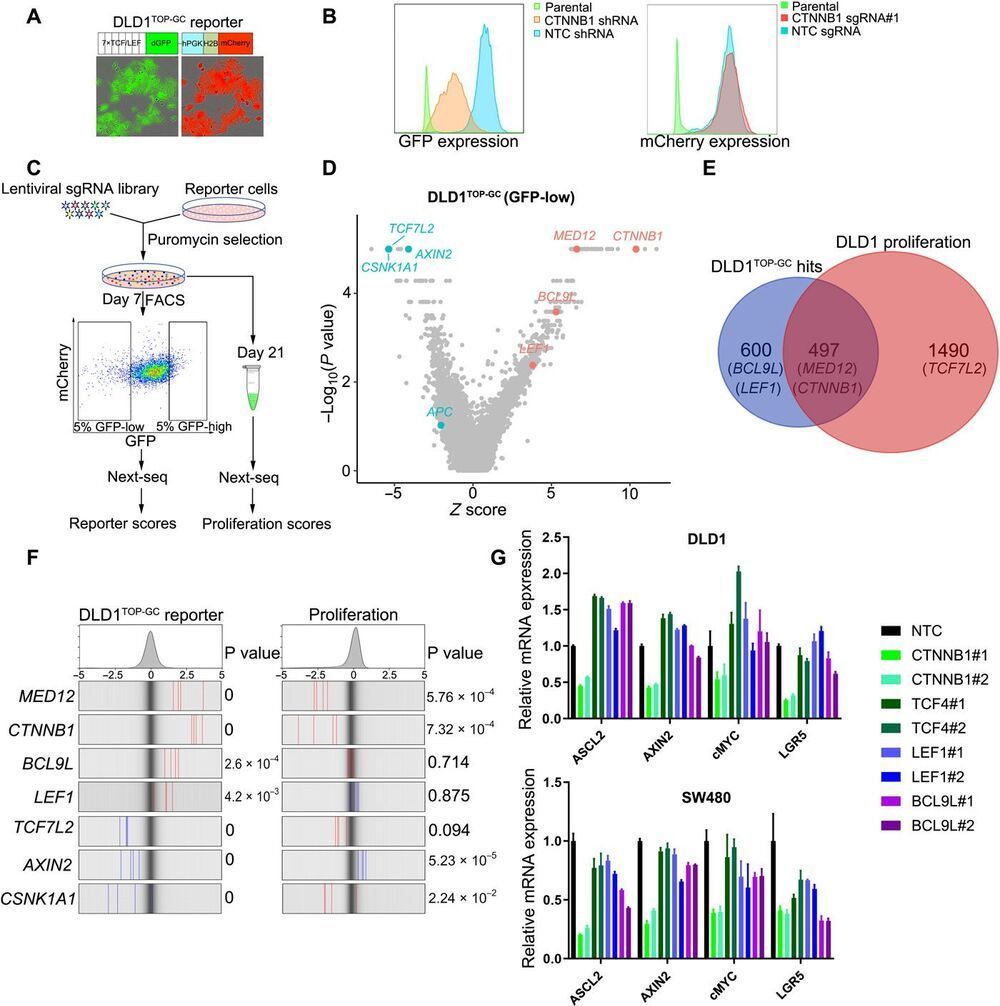
Nearly all colorectal tumors (CRC) harbor genetic mutations that lead to the hyperactivation of β-catenin signaling. For example, germline or spontaneous mutations in tumor suppressor APC may cause constitutive activation of β-catenin in colon stem cells and the development of colonic polyps, which may eventually evolve into colorectal carcinomas. Hyperactivated β-catenin initiates the expression of various downstream targets through binding to the promoter regions via TCF/LEF transcription factors. Studies using transcriptomic approaches have characterized various β-catenin–responsive targets, such as cMYC, AXIN2, ASCL2, LGR5, and CD44. Collectively, these targets promote proliferation (e.g., cMYC) and maintain a stem cell state (e.g., LGR5 and ASCL2), highlighting the potential value of developing treatments that target β-catenin signaling in cancer. However, β-catenin itself is an intractable drug target (6, 7).