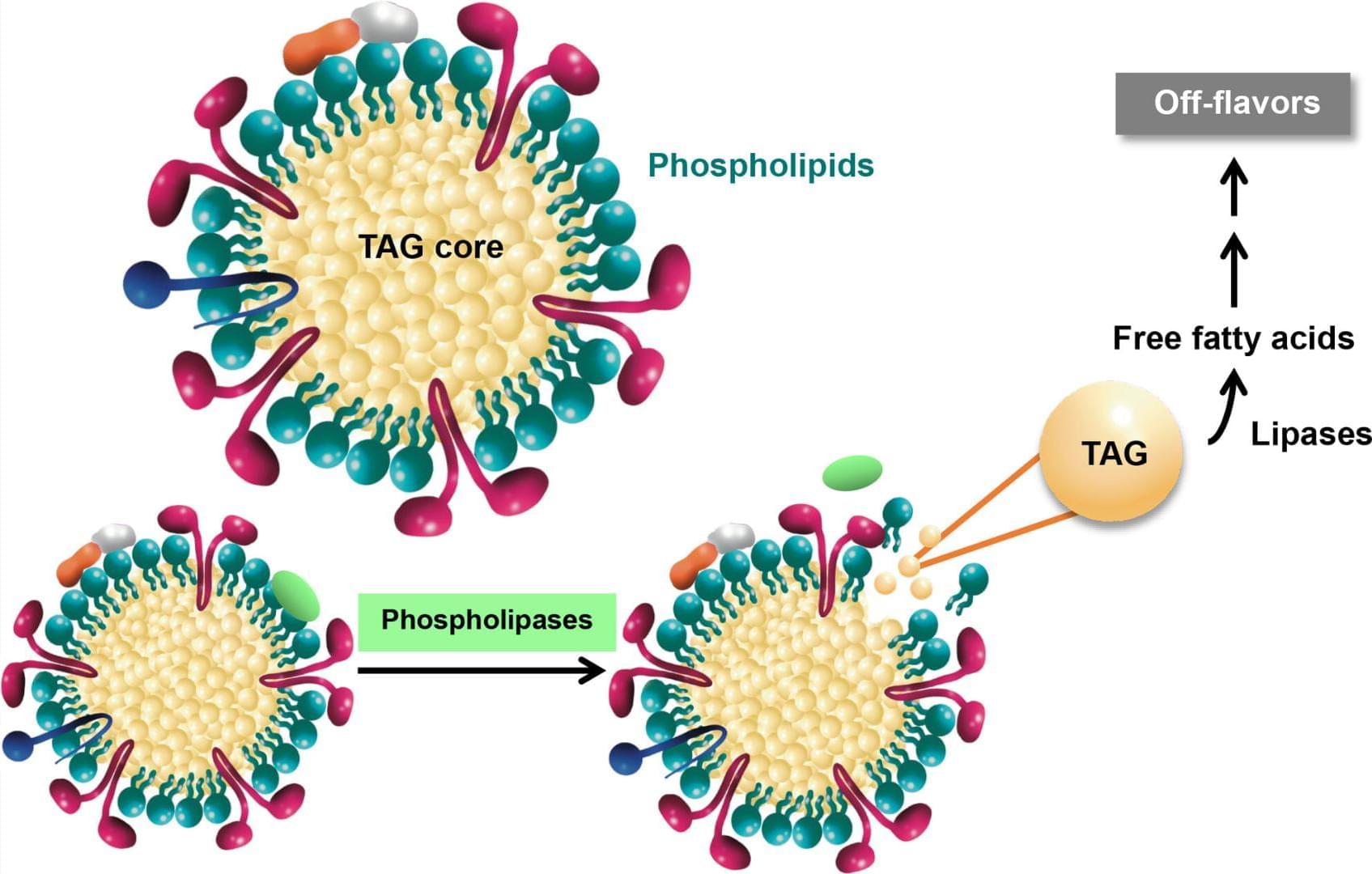
Schematic overview of phospholipase-mediated phospholipid hydrolysis and its impact on triacylglycerol (TAG) stability during rice bran storage.
Pancreatic ductal adenocarcinoma, the most common form of pancreatic cancer, is particularly deadly and hard to treat.
Most tumors of this type are driven by one or more mutations in the KRAS gene, pushing rapid cell division that’s difficult to stop.
They’ve long been considered so challenging to treat that KRAS has been labeled “undruggable” across decades of prior research.
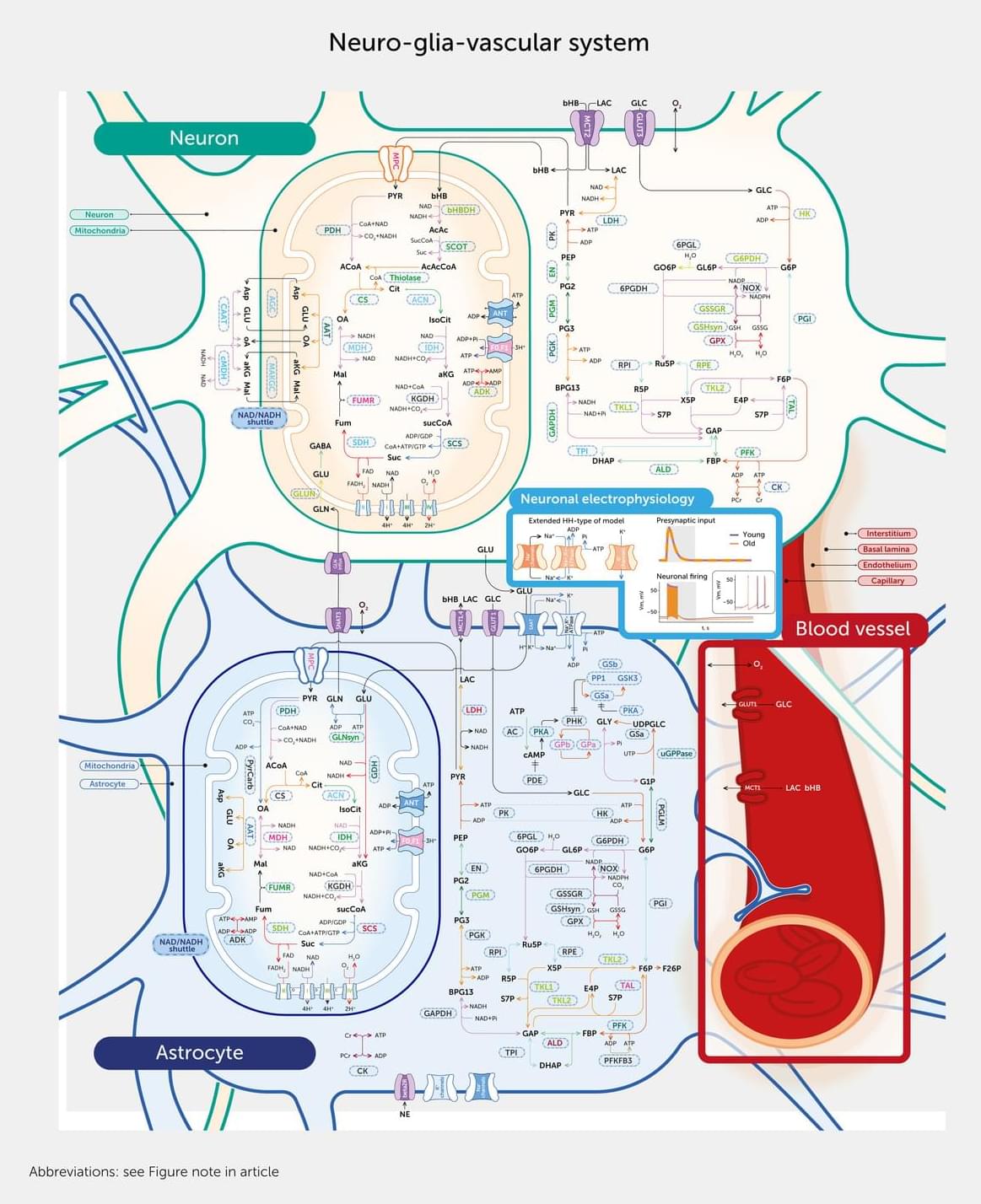
Age-related neurodegenerative disorders, including dementia, are a major global health concern. This article describes the first comprehensive, data-driven molecular model of the neuro-glia-vascular system to explore the complex relationships between the aging brain, energy metabolism, blood flow, and neuronal activity. Comprising 16,800 interaction pathways, the model includes all key enzymes, transporters, metabolites, and circulatory factors vital for neuronal electrical activity. We found significant alterations in metabolite concentrations and differential effects on adenosine triphosphate (ATP) supply in neurons and astrocytes and within subcellular compartments in aged brains and identified reduced sodium/potassium adenosine triphosphatase (Na+/K+-ATPase) activity as the leading cause of impaired neuronal action potentials.

🧬 What if every human had their own Digital Twin?
Not in 100 years.
Not in science fiction.
But within our lifetime.
For the past months, I’ve been building DiGem — a project focused on creating a Human Digital Twin: a digital representation of a person that combines health data, AI, lifestyle habits, and gamification into one system.
Imagine:
⚡ Your body displayed as a dashboard 🧠 AI acting as your personal health coach 📈 Real-time monitoring of your health and performance 🎮 Improving yourself through levels, XP, and achievements 🧬 A digital twin that evolves together with you.
On Sunday, July 5, 2026, at 1 p.m. U.S. Pacific Time, watch a compilation stream of four additional presentations from the May 2–3, 2026, sessions at the University of California, Berkeley Conference on Aging and Longevity (BerkeleyCAL), hosted by Professor Steven A. Garan, Director of Bioinformatics at the Center for Research and Education on Aging.
These presentations focus key insights in geroscience, both from its history and in regard to promising future directions and some implications for effective advocacy; they are delivered by some of the leading researchers in longevity science – Aubrey de Grey, Brendan Hughes, Felipe Sierra, and Michael West. Three of the presentations include question-and-answer sessions.
Dr. Aubrey de Grey of the Longevity Escape Velocity Foundation (LEVF) discusses the historical approaches to viewing aging and their shortcomings, as well as the damage-repair approach that he has championed and its prospects for rejuvenating the body. He also discusses implications for advocacy and which tactics could be more effective in bringing the public on board. Note: This presentation is an excerpt, captured by USTP Chairman Stolyarov on his phone camera. It is being made available due to the official recording having been lost.
Dr. Brendan Hughes from the Buck Institute discusses his thesis research on how DNA damage shapes unique, disease-relevant senescent cell states in neurons and other brain cell types. He details a methodology involving the direct differentiation of fibroblasts into neurons and oligodendrocytes to better understand aging-related cellular responses and potential therapeutic targets for Alzheimer’s disease. Dr. Hughes also highlights the importance of basic research in developing future interventions, such as senescence-targeted therapies or DNA repair modulations. The question-and-answer session includes a question from USTP Chairman Stolyarov to Dr. Hughes.
Dr. Felipe Sierra advocates for a shift in geroscience from solely targeting age-related diseases to focusing on maintaining intrinsic health and functional capacity. He proposes that molecular resilience acts as the crucial link between aging biology and long-term health, suggesting that strengthening this resilience could prevent the onset of multiple morbidities. Ultimately, he calls for more robust longitudinal studies and clinical trials that prioritize health-span metrics over the traditional, disease-centered approach to geriatric medicine.
Dr. Michael West explores the biological dichotomy between mortal somatic cells and the immortal germline to explain the fundamental mechanisms of aging and cellular regeneration. He discusses the history of stem-cell research and his work on telomeres and nuclear transfer, which demonstrated that developmental aging and cellular differentiation are reversible processes. Dr. West proposes a new approach to regenerative medicine that focuses on unlocking the body’s innate potential by targeting heterochrony genes to combat chronic degenerative diseases.
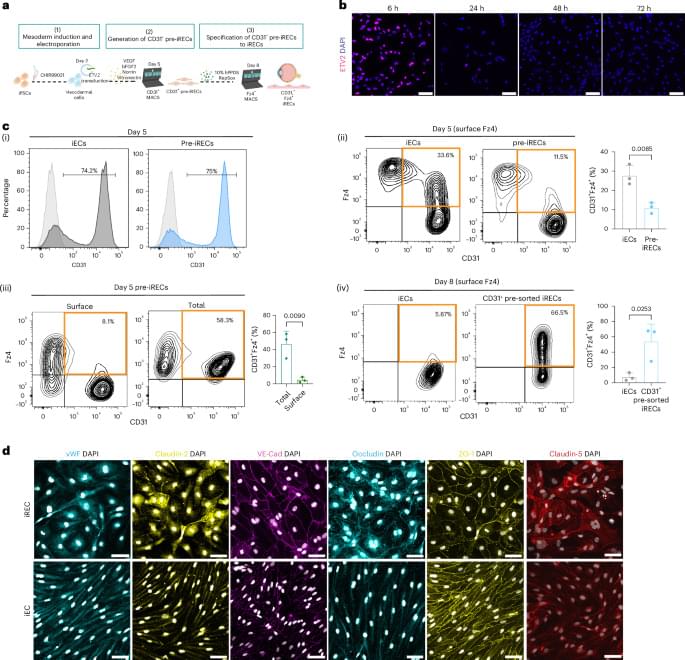
Treatment of induced pluripotent stem cells with Norrin and vitronectin, and subsequent cell sorting and maturation, generates retinal-like endothelial cells that can regenerate retinal vasculature in an oxygen-induced retinopathy mouse and recapitulate enhanced inner blood–retinal barrier properties when co-cultured with retinal-like pericytes.
Join us on Patreon! / michaellustgartenphd.
Discount Links/Affiliates:
Blood testing (where I get the majority of my labs, for those who blood test with Quest): https://www.ultalabtests.com/partners… those who blood test with LabCorp: https://www.anrdoezrs.net/click-10161… At-Home Metabolomics: https://www.iollo.com?ref=michael-lus… Use Code: CONQUERAGING At Checkout Clearly Filtered Water Filter: https://get.aspr.app/SHoPY Epigenetic, Telomere Testing: https://trudiagnostic.com/?irclickid=… Use Code: CONQUERAGING NAD+ Quantification: https://www.jinfiniti.com/intracellul… Use Code: ConquerAging At Checkout Oral Microbiome: https://www.bristlehealth.com/?ref=mi… Enter Code: ConquerAging SiphoxHealth Blood Testing (ApoB, GrimAge): https://siphoxhealth.com/mlustgarten Green Tea: https://www.ochaandco.com/?ref=fqbtflod Use Code: ML10OFF Diet Tracking: https://shareasale.com/r.cfm?b=139013… If you’d like to support the channel, you can do that with the website, Buy Me A Coffee: https://www.buymeacoffee.com/mlhnrca Conquer Aging Or Die Trying Merch! https://my-store-d4e7df.creator-sprin…
Blood Testing Essentials (Biological Age, CVD-Risk, Kidney Health and Function):
PhenoAge (Biological Age): https://www.ultalabtests.com/partners…
Measure the Bortz biological clock biomarkers: https://www.ultalabtests.com/partners…
Calculate your biological age using the Bortz clock: https://www.longevity-tools.com/human…
“That which does not kill us only makes us stranger.”
14 years ago, I sat down with Dr. Anders Sandberg, computational neuroscientist and research fellow at Oxford’s Future of Humanity Institute, for his second appearance on my podcast. His twist on Nietzsche has stayed with me ever since.
This was 2012. Before ChatGPT, before CRISPR babies, before Neuralink implants in human skulls. And yet listen to what we covered:
The ethics of transhumanism and the limits of being human The Epic of Gilgamesh and humanity’s oldest obsession: immortality Enhancement arms-races and the risk of conflict between transhumanists and neo-luddites Hive-minds, distributed intelligence, and whether the Borg should scare us Mind uploading and what survives when the body doesn’t.
What strikes me now, rewatching it, is how little the fundamental questions have changed. The technology raced ahead. The philosophy is still catching up.
Anders argued that embracing strangeness is not a bug of the human future; it’s the feature. The question was never whether we would change. It’s whether we will change wisely.
{kind=link}