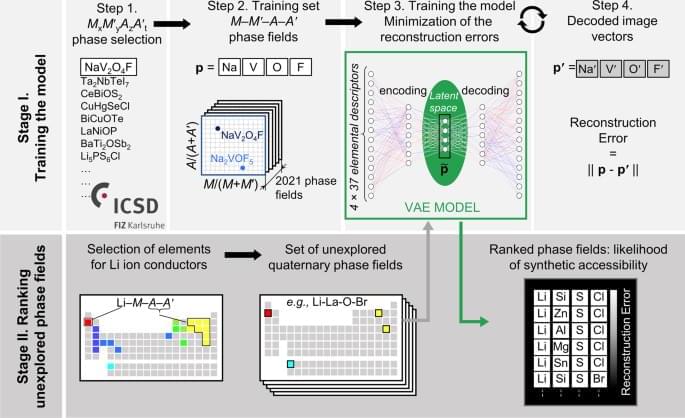
Machine learning (ML) models are powerful tools to study multivariate correlations that exist within large datasets but are hard for humans to identify16,23. Our aim is to build a model that captures the chemical interactions between the element combinations that afford reported crystalline inorganic materials, noting that the aim of such models is efficacy rather than interpretability, and that as such they can be complementary guides to human experts. The model should assist expert prioritization between the promising element combinations by ranking them quantitatively. Researchers have practically understood how to identify new chemistries based on element combinations for phase-field exploration, but not at significant scale. However, the prioritization of these attractive knowledge-based choices for experimental and computational investigation is critical as it determines substantial resource commitment. The collaborative ML workflow24,25 developed here includes a ML tool trained across all available data at a scale beyond that, which humans can assimilate simultaneously to provide numerical ranking of the likelihood of identifying new phases in the selected chemistries. We illustrate the predictive power of ML in this workflow in the discovery of a new solid-state Li-ion conductor from unexplored quaternary phase fields with two anions. To train a model to assist prioritization of these candidate phase fields, we extracted 2021 MxM ′yAzA ′t phases reported in ICSD (Fig. 1, Step 1), and associated each phase with the phase fields M-M ′-A-A′ where M, M ′ span all cations, A, A ′ are anions {N3−, P3−, As3−, O2−, S2−, Se2−, Te2−, F−, Cl−, Br−, and I−} and x, y, z, t denote concentrations (Fig. 1, Step 2). Data were augmented by 24-fold elemental permutations to enhance learning and prevent overfitting (Supplementary Fig. 2).
ML models rely on using appropriate features (often called descriptors)26 to describe the data presented, so feature selection is critical to the quality of the model. The challenge of selecting the best set of features among the multitude available for the chemical elements (e.g., atomic weight, valence, ionic radius, etc.)26 lies in balancing competing considerations: a small number of features usually makes learning more robust, while limiting the predictive power of resulting models, large numbers of features tend to make models more descriptive and discriminating while increasing the risk of overfitting. We evaluated 40 individual features26,27 (Supplementary Fig. 4, 5) that have reported values for all elements and identify a set of 37 elemental features that best balance these considerations. We thus describe each phase field of four elements as a vector in a 148-dimensional feature space (37 features × 4 elements = 148 dimensions).
To infer relationships between entries in such a high-dimensional feature space in which the training data are necessarily sparsely distributed28, we employ the variational autoencoder (VAE), an unsupervised neural network-based dimensionality reduction method (Fig. 1, Step 3), which quantifies nonlinear similarities in high-dimensional unlabelled data29 and, in addition to the conventional autoencoder, pays close attention to the distribution of the data features in multidimensional space. A VAE is a two-part neural network, where one part is used to compress (encode) the input vectors into a lower-dimensional (latent) space, and the other to decode vectors in latent space back into the original high-dimensional space. Here we choose to encode the 148-dimensional input feature space into a four-dimensional latent feature space (Supplementary Methods).