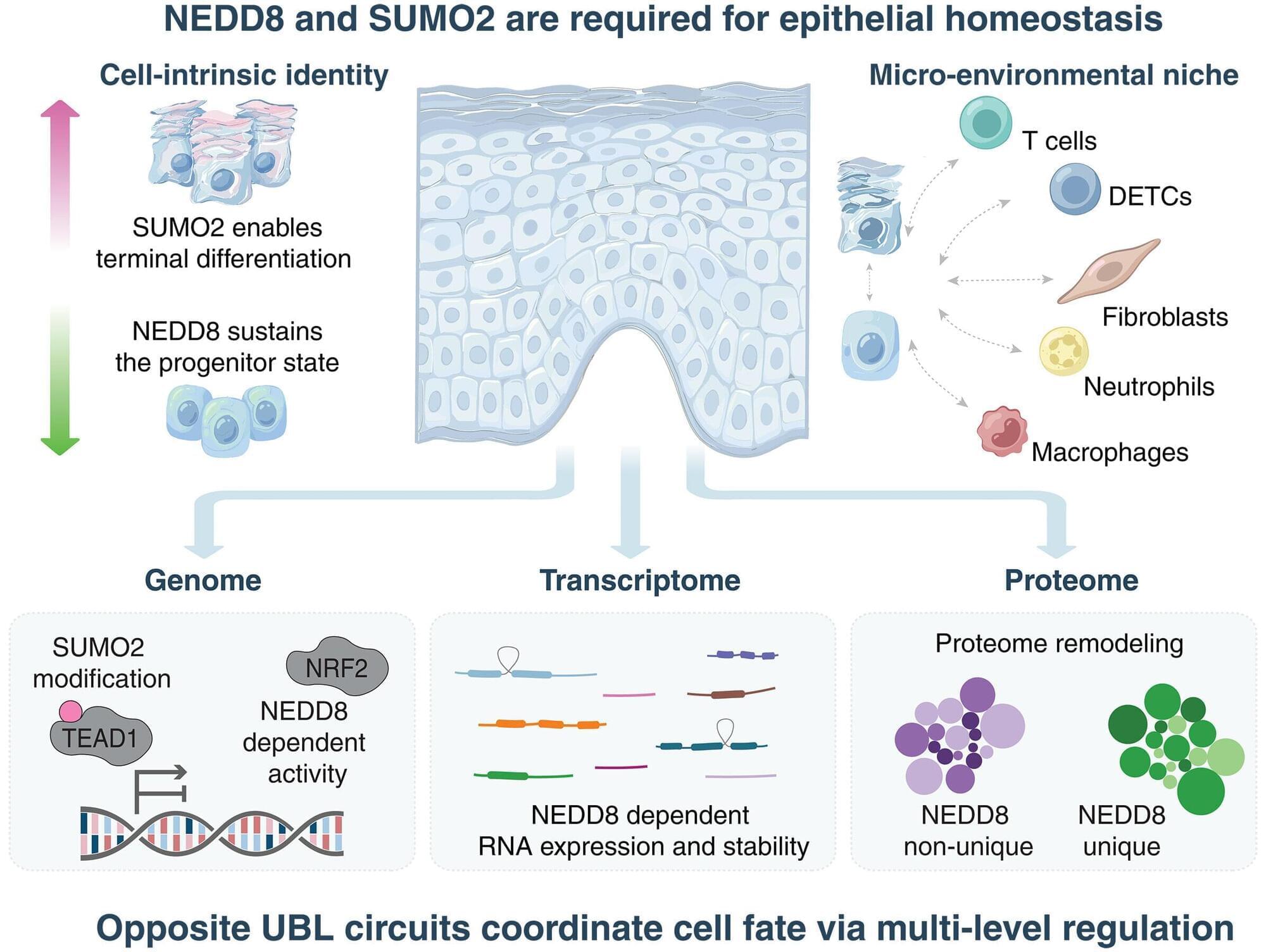
Two proteins with opposing functions orchestrate the development and maintenance of healthy skin, Stanford Medicine researchers have found. Modulating their activity with topical drugs could reduce inflammation, aid wound healing and slow or halt the growth of skin cancer, the researchers believe. The findings are published in the journal Science.
The proteins are part of a family called ubiquitin-like proteins. Ubiquitination controls the targeted destruction and disposal of unneeded proteins in a cell. But in the skin, certain ubiquitin-like proteins instead switch on or off wide swaths of genes involved in cellular growth and development, the study found. In particular, they trigger progenitor (stem) cells in the lower layer of the skin to either mature and migrate to the skin surface or to self-renew.
“These two ubiquitin-like protein systems are remarkably dedicated and opposite in their functions,” said Paul Khavari, MD, Ph.D., chair of dermatology at the Stanford School of Medicine and senior author of the study. “One promotes the stem-cell state while the other drives differentiation. It’s like having two opposing forces that determine a cell’s fate.”