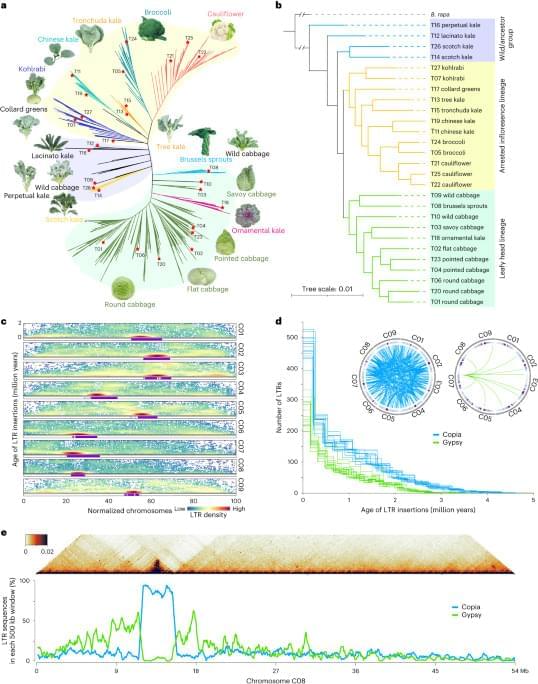
To construct a pan-genome that encompasses the full range of genetic diversity in B. ole racea, we analyzed the resequencing data of 704 globally distributed B. ole racea accessions covering all different morphotypes and their wild relatives (Supplementary Tables 1 and 2). We identified 3,792,290 SNPs and 528,850 InDels in these accessions using cabbage JZS as reference genome22. A phylogenetic tree was then constructed using SNPs, which classified the 704 accessions into the following three main groups: wild B. ole racea and kales, arrested inflorescence lineage (AIL) and leafy head lineage (LHL; Fig. 1a and Supplementary Note 2). The phylogenetic relationship revealed in our study was generally consistent with those reported previously4,5,24,25. Based on the phylogeny and morphotype diversity, we selected 22 representative accessions for de novo genome assembly (Table 1).
We assembled genome sequences of the 22 accessions by integrating long-reads (PacBio or Nanopore sequencing), optical mapping molecules (BioNano) or high-throughput chromosome conformation capture data (Hi-C) and Illumina short-reads (Methods; Supplementary Note 2 and Supplementary Tables 3–7). The total genome size of these assemblies ranged from 539.87 to 584.16 Mb with an average contig N50 of 19.18 Mb (Table 1). An average of 98% contig sequences were anchored to the nine pseudochromosomes of B. ole racea. The completeness of these genome assemblies was assessed using benchmarking universal single-copy orthologs (BUSCO), with an average of 98.70% complete score in these genomes (Supplementary Table 8).
To minimize artifacts that could arise from different gene prediction approaches, we predicted gene models of both the 22 newly assembled genomes and the five reported high-quality genomes5,21,22,23 using the same annotation pipeline (Methods). Using an integrated strategy combining ab initio, homology-based and transcriptome-assisted prediction, we obtained a range of 50,346 to 55,003 protein-coding genes with a mean BUSCO value of 97.9% in these genomes (Table 1). After gene prediction, a phylogenetic tree constructed based on single-copy orthologous genes clustered the 27 genomes into three groups, similar to the results observed in the population (Fig. 1a and b).