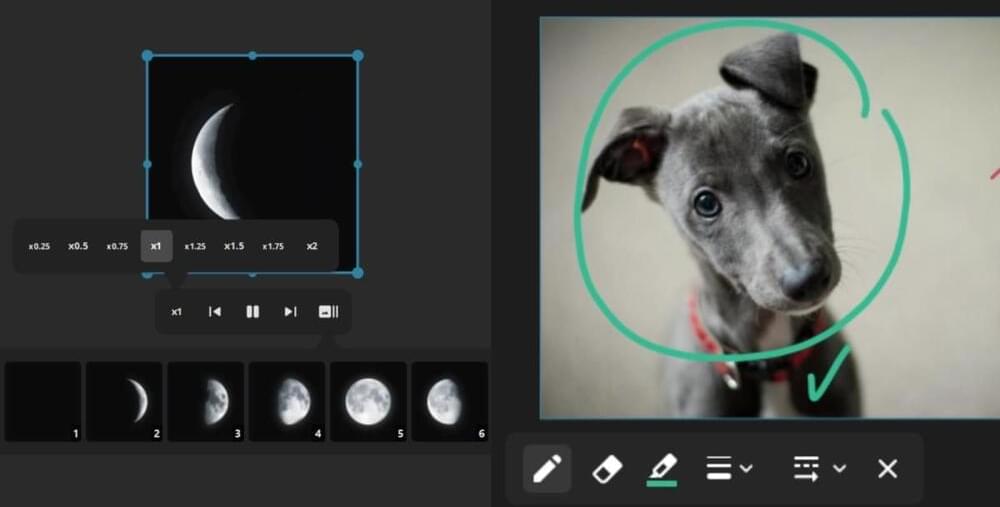
The long-awaited PureRef 2.0 update has arrived, introducing visual and performance improvements, as well as some of the most requested features.

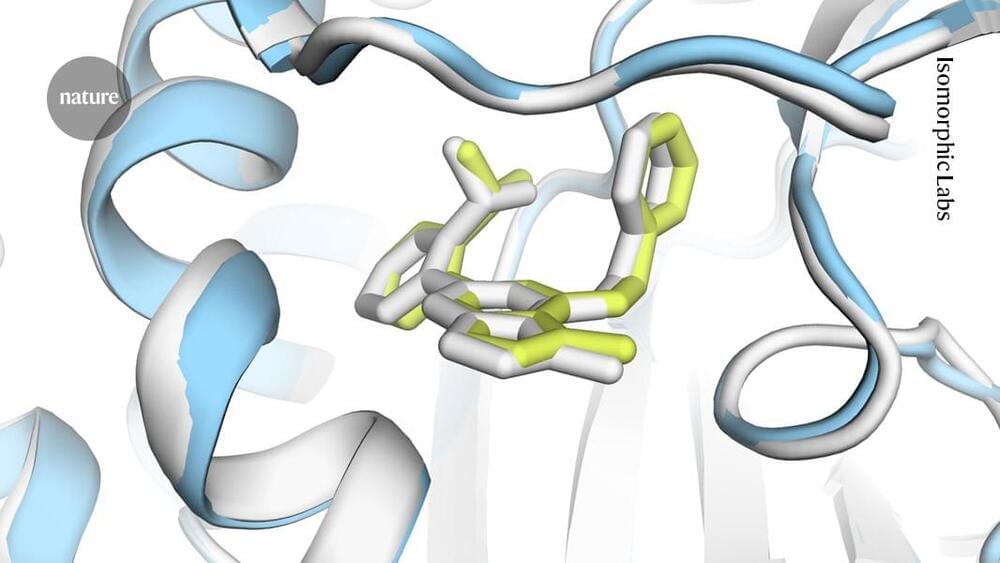
Recent experimental advancements have enabled more accurate and in-depth analysis of these materials during and after formation. The review article examines two decades of research on the non-classical formation pathways of soft and organic crystalline materials. It details the current theoretical understanding of how these materials form through non-classical pathways, including distinguishing the processes of nucleation and growth across models.
Advances in experimental methods, including in-line scattering/spectroscopy detection, cryo microscopy, and in situ liquid-phase characterization, and their application to studying soft and organic crystalline materials are also discussed.
These experimental techniques have provided strong evidence for non-classical crystallization pathways, leading to key breakthroughs in understanding these processes. However, the sole presence of a specific final product or intermediate does not prove that a material formed via a specific pathway.

Elon Musk, the billionaire CEO of Tesla, has said that artificial intelligence (AI) will eventually eliminate all jobs. However, he affirmed that this is not necessarily an adverse development.
Speaking at a startup and tech event in Paris on Thursday (May 23) via video link, Musk said, “Probably none of us will have a job”, while predicting a future where jobs would be “optional”
“If you want to do a job that’s kinda like a hobby, you can do a job,” Musk told VivaTech 2024. “But otherwise, AI and the robots will provide any goods and services that you want.”



Join our newsletter to get the latest military space news every Tuesday by veteran defense journalist Sandra Erwin.
In a statement May 22, the Space Force said this specialized environment will be crucial for training service personnel, known as guardians, to defend critical satellites and other spacecraft from electronic attacks. Satellites rely on electromagnetic signals for communication, navigation, and data transmission, making them vulnerable to jamming and cyberattacks.

Jamming and spoofing attacks on GPS and other global navigation satellite systems (GNSS) are becoming increasingly common as geopolitical crises escalate, creating major challenges and risks for aviation, shipping and other critical services across the world.
Data from GPSJam.org has confirmed widespread GPS/GNSS interference across parts of Europe and beyond as an outcome of the war in Ukraine. Regions affected range from Finland and the Baltics to Poland, Romania and Bulgaria — in addition to the Black Sea, the Caucasus and Turkey. The Middle East is also being affected by interference stemming from Israel and Iran’s hostile activities in the region. Other interference efforts, albeit at a lower scale, are also regularly occurring in areas of Pakistan, India and Myanmar.
This interference can cause significant disruptions to airline take-offs and landings, leading to costly flight delays and flight plan changes. It also presents real risks for certain aircraft and airports. For instance, some airports rely solely on GPS signals for their method of approach —– this is why Russian GPS jamming forced Finnair to suspend flights to Estonia’s Tartu Airport earlier this year. In 2019, a passenger aircraft in Idaho nearly crashed into a mountain due to GPS disruption.
{kind=link}